 浙江省粘接技术协会
浙江省粘接技术协会 张兴宏 叶胜荣
浙江大学高分子科学与工程系,高分子合成与功能构造教育部重点实验室 浙江杭州 310027,Email: xhzhang@zju.edu.cn
摘 要 非异氰酸酯路线合成聚氨酯是聚氨酯合成的新兴研究方向,可避免储运难、毒性大的异氰酸酯的使用。本文以双环氧树脂、环氧丙烷,CO2为起始原料,采用双金属氰化络合物(DMCC)为/十六烷基三甲基溴化铵催化体系催化“一锅”反应,100%选择性地合成得到碳酸丙烯酯和多环碳酸酯化合物。反应过程中生成的碳酸丙烯酯也起到环氧树脂环化反应的良溶剂,有效地促进了双环氧树脂环氧基团100%转化为环碳酸酯。研究结果为非异氰酸酯路线提供了制备新结构的多环碳酸酯新的合成方法。
关键词 双金属氰化络合物 聚氨酯 非异氰酸酯路线
1 引言
聚氨酯(Polyurethane, PU)是一种重要的高分子材料,具有高强度、高耐磨和耐高、低温等优良性能,可制成弹性体、发泡材料、胶粘剂等,广泛用于汽车制造、合成革、石油化工、农业和交通运输等领域。传统聚氨酯的制备需用到有毒的化学物质异氰酸酯,而异氰酸酯化学性质活泼、储运不便,同时异氰酸酯一般是由剧毒的光气制造而来,潜在的环境危害巨大。因此,随着社会发展和人们对环境的日益关注,这种传统制备聚氨酯的方法逐渐受到约束和限制,因此发展一种环境友好,安全卫生的非异氰酸酯方法来制备聚氨酯成为关注的热点,其中,以二氧化碳(CO2)为原料与环氧化合物偶合反应合成多环碳酸酯、再用环碳酸酯与二元胺制备聚氨酯这一途径日益受到人们的广泛关注[1-4]。
二氧化碳(CO2)被认为是一种大气“污染物”,是导致温室效应和全球变暖的罪魁祸首。但从合成角度看,它是一种丰富易得的C1原料。因此从CO2出发直接合成有用的化学物质,既实现化学固定CO2并有效利用,又有利于CO2的减排,对推动经济、社会的可持续发展具有重要意义[5]。其中,CO2与环氧化物偶合反应制备环碳酸酯是一种极具前景的方法。这是因为反应产物环碳酸酯是一种非常重要的极性非质子溶剂和反应中间体。但迄今,多官能团环氧与CO2偶合反应的报道很少。一些报道[6-7]在极性有机溶剂如四氢呋喃,二甲基亚砜,N-甲基吡咯烷酮存在下,用均相催化剂卤化锂催化二(多)环氧化合物如双酚A二缩水甘油醚,聚(丙烯酸缩水甘油醚)等与CO2偶合反应,可以100%高选择性的合成制备相应的环碳酸酯,然后合成的二元(多元)环碳酸酯再与不同结构二元胺反应,可以得到结构不同、性能各异的聚氨酯[6-7]。
催化二环氧化合物与CO2偶合反应,由于二环氧化合物与单环氧化合物如环氧丙烷相比,单体粘度较大,不利于催化剂的分散,一般需要在反应体系中外加极性有机溶剂,以促进反应的进行。在大部分报道中,外加的有机溶剂如四氢呋喃,二甲基亚砜,二氧六环和N-甲基吡咯烷酮一般都有或大或小的毒性,且反应后脱除麻烦,也不符合环境友好的要求。针对这一问题,本文采用双金属氰化络合物(double metal cyanide complex, DMCC)/十六烷基三甲基溴化铵 (CTAB)二元催化体系,直接催化环氧丙烷(propylene oxide, PO)、二环氧单体和CO2“一锅”法高选择性合成碳酸丙烯酯和二环碳酸酯两种产物。其中,生成的碳酸丙烯酯可以作为多环氧单体偶合反应的良溶剂。该催化体系相较于已经报道的均相卤化锂催化剂,可以同时100%高选择性地催化PO/CO2偶合反应和二环氧化合物/CO2偶合反应制备相应的碳酸丙烯酯和二元环碳酸酯,单体的转化率均近100%,且催化剂用量小。本工作为非异氰酸酯路线提供了合成不同结构的多环碳酸酯单体的制备方法,所合成的多环碳酸酯及相应的非异氰酸酯路线具有较好的工业化前景。
2. 实验部分
2.1 材料
K3Co(CN)6(ACROS,95%)使用前用热水重结晶。ZnCl2,叔丁醇(t-BuOH),十六烷基三甲基溴化铵,四氢呋喃,二氯甲烷,二甲基亚砜,N-甲基吡咯烷酮,二氧六环,碳酸丙烯酯均为分析纯试剂,未经纯化直接使用。环氧丙烷(PO)用氢化钙(CaH2)浸泡12h,然后回流4小时候得到。环氧单体有结晶性双酚A二缩水甘油醚、环己二醇二缩水甘油醚、3,3,5,5-四甲基联苯双酚二缩水甘油醚、丁二醇二缩水甘油醚、5,5-二甲基海因二缩水甘油醚和新戊二醇二缩水甘油醚,均为工业品,未经纯化直接使用。
2.2 DMCC催化剂的制备
按照本实验室文献方法合成[8-9]。
2.3环碳酸酯的合成
将带磁力搅拌的100mL高压反应釜用四氢呋喃溶剂反复清洗干净,然后80℃下真空干燥5小时。将设定量DMCC催化剂、十六烷基三甲基溴化铵溴化铵和环氧单体加入至高压反应釜中,继续60℃真空干燥3小时,然后冷却至室温,在负压条件下用注射器向釜内加入一定体积的有机溶剂或者PO。然后向釜内压入一定压力CO2,升高反应温度至设定温度时,调节釜内压力至设定值。整个反应过程全程搅拌,搅拌速率为500r/min。反应结束以后,用冰水浴冷却至室温,缓慢排出未反应CO2,粗产物通过减压蒸馏除去有机溶剂和未反应的单体(PO)后进行1H NMR表征。对于结晶性双酚A二缩水甘油醚和3,3,5,5-四甲基联苯双酚二缩水甘油醚,其环碳酸酯产物可用四氢呋喃过滤洗涤,除去未反应单体,对于其它几种选用的二环氧单体及相应产物,先用丙酮溶解,然后用饱和的氯化钠水溶液沉淀,过滤,除水以后得到相应的环碳酸酯,真空干燥,称重,计算转化率。
3. 结果与讨论
3.1 溶剂对双环氧树脂/CO2偶合反应的影响。
由于双环氧树脂如双酚A二缩水甘油醚等常温下粘性较大,其与CO2的偶合反应中需额外添加有机溶剂,以促进催化剂的分散和热交换,提高反应速率。本文首先采用双酚A二缩水甘油醚与CO2反应,在15mg双金属氰化络合物催化剂、150mg季铵盐、4MPa和120℃条件下,加入不同的有机溶剂,考察不同有机溶剂对双环氧树脂转化率的影响。结果如表1所示。当采用二氧六环或二甲基亚砜为溶剂时,环氧基团的转化率最低,仅为52.9%和58.1%,而使用碳酸丙烯酯时,表现出最高的转化率,达到84.1%,其它溶剂如四氢呋喃、二氯甲烷和N-甲基吡咯烷酮则居于两者之间。不同溶剂导致双环氧树脂转化率不同的原因可能与十六烷基三甲基溴化铵在各溶剂中的溶解度和DMCC在溶剂中的易分散性有关。
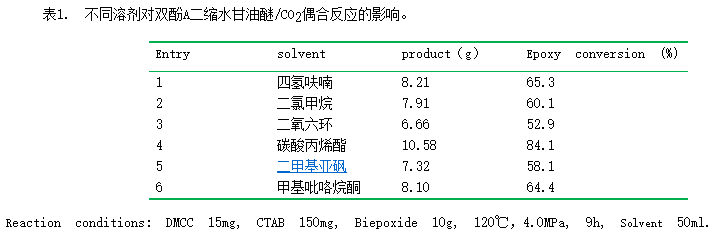
3.2 反应条件对双环氧树脂/CO2偶合反应的影响
以四氢呋喃为溶剂,考察DMCC催化剂/季铵盐配比、反应时间和压力对双酚A二缩水甘油醚/CO2偶合反应的影响,结果列于表2。分别单独使用DMCC与十六烷基三甲基溴化铵时,两者均能催化双环氧树脂/CO2偶合反应(表2,entries1和6)。但DMCC的催化活性非常低,而季铵盐则表现出40.0%的环氧基团转化率。当两者结合使用时,环氧基团转化率可显著提高至65.9%。这表明DMCC与季铵盐之间存在协同催化作用,有利于双环氧树脂环氧基团转化率的增加。我们推测两者协同催化机理是:首先环氧基团上的氧原子与DMCC催化剂中的金属活性中心配位,促使环氧基活化,然后季铵盐中的溴阴离子作为一个有效的亲核试剂,进攻环氧基团上位阻较小的碳原子,促使环氧基开环,从而有利于CO2的插入。
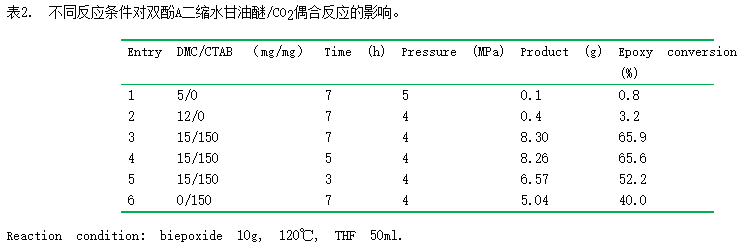
图1是双酚A二缩水甘油醚/CO2偶合产物的1H NMR谱图。由图可见,环碳酸酯基团上三个氢原子的化学位移峰分别在5.1ppm、4.56ppm和4.23ppm处,而在谱图中并未发现环氧基团上氢原子的相关化学位移。
图 1. DMCC/CTAB催化双酚A二缩水甘油醚/CO2偶合反应产物的1H NMR谱图。
3.3 环氧丙烷/双环氧树脂/CO2一锅法制备碳酸丙烯酯与双环碳酸酯树脂
由表1可知,当使用碳酸丙烯酯为有机溶剂时,双酚A二缩水甘油醚的环氧基团的转化率最高。考虑到碳酸丙烯酯可由环氧丙烷与CO2偶合反应制备,因此我们希望使用DMCC/十六烷基三甲基溴化铵催化体系一步催化环氧丙烷/双环氧树脂/CO2反应,同时高选择性获得碳酸丙烯酯与双环碳酸酯两种产物。实验结果如表3所示。
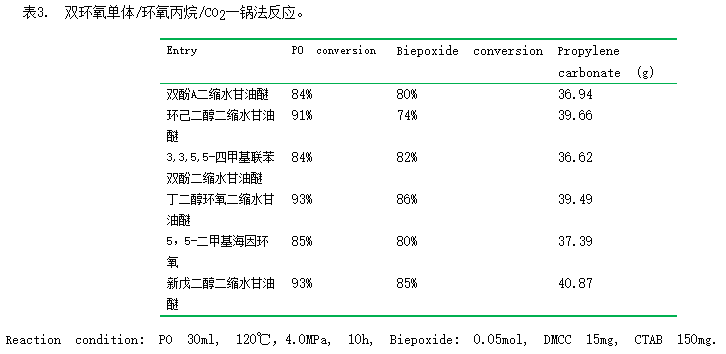
所采用的环氧树脂有双酚A二缩水甘油醚、环己二醇二缩水甘油醚、3,3,5,5-四甲基二缩水甘油醚、丁二醇二缩水甘油醚、5,5-二甲基环氧和新戊二醇二缩水甘油醚。由表3可见,在“一锅”反应中,除环己二醇二缩水甘油醚转化率相对较低外,其它几种双环氧单体的转化率均达到80%以上,反应体系中环氧丙烷的转化率最高可达93%。说明该二元催化体系可以高活性、高选择性的同时制备碳酸丙烯酯和二元环碳酸酯两种产物。图2-7分别表中双环氧单体与CO2偶合反应的产物二环碳酸酯产物的1H NMR谱图。从谱图中可见,环氧单体中的环氧基团上的氢原子的化学位移(2-3ppm之间),经过偶合反应之后,这些峰消失,而在4-5ppm范围内出现了二元环碳酸酯的特征质子氢的化学位移,说明该催化体系成功制备了二环碳酸酯。经过分离后,所得的二环碳酸酯纯度很高,从NMR谱图上未见环氧单体的特征峰。说明经过洗涤后,部分未反应的二环氧化物、或一端反应成为环碳酸酯的环氧化物均能除去,这为下一步与二胺反应制备聚氨酯奠定了基础。
4. 结论
采用双金属氰化络合物(DMCC)/十六烷基三甲基溴化铵二元催化体系,催化环氧丙烷/双环氧树脂/CO2一锅反应,高活性高选择性制备了碳酸丙烯酯与二环碳酸酯两种环碳酸酯。反应过程中生成的碳酸丙烯酯可为二环碳酸酯的的溶剂,从而保证了高选择性、高活性生成二元环碳酸酯。由于后者可通过与二胺的反应制备含羟基官能团的聚氨酯,因此本文提供了一种高效高选择性合成多环碳酸酯的方法,为非异氰酸酯路线制备聚氨酯奠定了基础。
参考文献:
(1) McCabe RW, Taylor A. Chem. Commun. 2002, 934.
(2) Rokicki G, Piotrowska A. Polymer 2002, 43:2927.
(3) Ubaghs L, Fricke N, Keul H, Hocker H. Macromol. Rapid Commun. 2004, 25:517.
(4) Kihara N, Kushida Y, Endo T. J. Polym. Sci. Part A: Polym. Chem. 1996, 34:2173.
(5) Yoshida M and Ihara M. Chem. Eur. J. 2004, 10:2886.
(6) Ochiai B, Inoue S, Endo T. J. Polym. Sci. Part A: Polym. Chem. 2005, 43:6613.
(7) Ochiai B, Hatano Y, and Endo T. Macromolecules 2008, 41:9937.
(8) Chen S, Hua ZJ, Fang Z, Qi GR. Polymer 2004, 45: 6519.
(9) Zhang XH, Wei RJ, Sun XK, Zhang ZF, Du BY, Fan ZQ, Qi GR. Polymer 2011, 52: 5494.




